Home
My projects
Sometimes I create stuff
biosubg
Directed random walks and constraint programming reveal active pathways in hepatocyte growth factor signaling
An effective means to analyze mRNA expression data is to take advantage of established knowledge from pathway databases, using methods such as pathway‐enrichment analyses. However, pathway databases are not case‐specific and expression data could be used to infer gene‐regulation patterns in the context of specific pathways. In addition, canonical pathways may not always describe the signaling mechanisms properly, because interactions can frequently occur between genes in different pathways. Relatively few methods have been proposed to date for generating and analyzing such networks, preserving the causality between gene interactions and reasoning over the qualitative logic of regulatory effects.
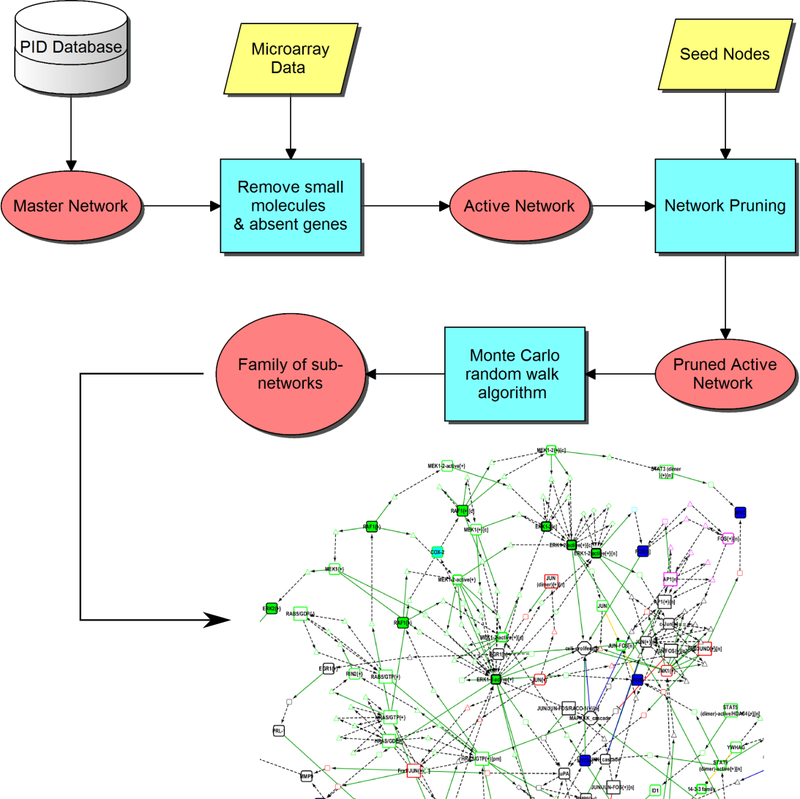 MCWalk is an algorithm integrated with a logic programming approach to discover subgraphs in large‐scale signaling networks by random walks in a fully automated pipeline. As an exemplary application, we used it to uncover the signal transduction mechanisms in a gene interaction network describing hepatocyte growth factor‐stimulated cell migration and proliferation from gene‐expression measured with microarray and RT‐qPCR using in‐house perturbation experiments in a keratinocyte‐fibroblast co‐culture. The resulting subgraphs illustrate possible associations of hepatocyte growth factor receptor c‐Met nodes, differentially expressed genes and cellular states. Using perturbation experiments and Answer Set programming, we are able to select those which are more consistent with the experimental data. We discover key regulator nodes by measuring the frequency with which they are traversed when connecting signaling between receptors and significantly regulated genes and predict their expression‐shift consistently with the measured data.
MCWalk is an algorithm integrated with a logic programming approach to discover subgraphs in large‐scale signaling networks by random walks in a fully automated pipeline. As an exemplary application, we used it to uncover the signal transduction mechanisms in a gene interaction network describing hepatocyte growth factor‐stimulated cell migration and proliferation from gene‐expression measured with microarray and RT‐qPCR using in‐house perturbation experiments in a keratinocyte‐fibroblast co‐culture. The resulting subgraphs illustrate possible associations of hepatocyte growth factor receptor c‐Met nodes, differentially expressed genes and cellular states. Using perturbation experiments and Answer Set programming, we are able to select those which are more consistent with the experimental data. We discover key regulator nodes by measuring the frequency with which they are traversed when connecting signaling between receptors and significantly regulated genes and predict their expression‐shift consistently with the measured data.
Read the full article here. The Java implementation of MCWalk is publicly available under the MIT license here.